Ongoing Projects
 DarkTree — Charting the dark regions of the insect tree using computer vision, genomics, and probabilistic machine learning
DarkTree — Charting the dark regions of the insect tree using computer vision, genomics, and probabilistic machine learning
Systematic genome sequencing is opening up new research frontiers across the life sciences.
But how do you approach a hyperdiverse group like insects? We used to believe there were about
5.5 million insect species on Earth, only 20% of which had been described. However, recent
inventories using DNA metabarcoding suggest that the total diversity is in the tens of millions,
and that our knowledge of the insect tree of life is even more fragmented and biased than
previously thought. Current genome sequencing efforts tend to amplify rather than mitigate these
biases, seriously compromising the goal of obtaining a representative picture of insect evolution.
We will address the missing pieces in the insect genome tree using the unique global insect
material (60,000 samples, 30 million insects) collected by the Insect Biome Atlas and LIFEPLAN
projects. This project is a collaboration between the Swedish Museum of Natural History, KTH and
Linköping University, and launched in 2025.
Involved Researchers:
Elzbieta Iwaszkiewicz,
Claudia Weber,
Tim Virgoulay
 Finance to Revive Biodiversity (FinBio)
Finance to Revive Biodiversity (FinBio)
Biodiversity loss is unravelling the web of life that supports all people, societies, economies,
including the financial system. MISTRA Finance to Revive Biodiversity
(FinBio) will provide cutting-edge research to connect the
financial system and biodiversity. The program is hosted by Stockholm Resilience Centre at
Stockholm University and is a partnership between academic and financial actors. The mission is
to support the financial sector's capacity to contribute to a nature-positive economy, enhancing
the resilience of our planet by reversing the loss of nature and biodiversity.
The Ronquist lab will contribute by for example testing new environmental-DNA techniques to
track and map biodiversity change in ways that produce open source data to inform financial
decisions.
Involved Researchers:
Emma Granqvist,
Robert Goodsell
 TreePPL: A Probabilistic Programming Language for Phylogenetics and Biodiversity
TreePPL: A Probabilistic Programming Language for Phylogenetics and Biodiversity
This is a collaborative project with David Broman's group at KTH, developing a probabilistic
programming language for phylogenetics and biodiversity. More info at the project webpage:
TreePPL.
The project is supported by The Swedish Research Council (VR).
Involved Researchers:
Viktor Senderov,
Emma Granqvist,
Tim Virgoulay
Previous Projects
 Insect Biome Atlas (IBA)
Insect Biome Atlas (IBA)
Insect Biome Atlas (IBA) is an international
collaborative effort to describe in detail the insect faunas of two biologically and
geologically very different countries: Sweden and Madagascar. The project, one of the largest
ongoing insect biodiversity surveys, addresses key questions about the insect diversity: How
are insect species distributed across habitats, sites and seasons? What are the key
environmental factors shaping insect diversity?
Collection phase lasted a whole year (2019) in each country and was done by means of Malaise
traps: 200 in Sweden and 50 in Madagascar. Summing up, the IBA collected more than 8000 insect
community samples. Several other types of samples and ecological measurements were also
collected at the trap sites to gather a full understanding of the ecological roles of the
organisms that comprise the insect biome in these countries. Analysis of the samples include
identification of all insects and the organisms they interact with, such as pathogens as well
as symbiotic fungi and bacteria - this is achieved by using novel DNA techniques. The results
and material collected during the IBA project will be useful for scientists interested in
systematics, taxonomy and insect ecology and evolution for many years to come.
In Sweden the Malaise traps were managed by over 100 volunteers, which makes this project one
of the largest citizen science projects to take place in Scandinavia.
IBA consortium brings together researchers from many countries and institutes including
Natural History Museum in Stockholm,
Stockholm University,
Swedish University of Agricultural Sciences, Uppsala,
SciLifeLab, KTH Royal Institute of Technology
Stockholm, Madagascar Biodiversity Center,
Jagiellonian University, Krakow, Poland.
Involved Researchers:
Andreia Miraldo,
Laura van Dijk,
Robert Goodsell,
Emma Granqvist,
Elzbieta Iwaszkiewicz
 Cynipoid Phylogenomics
Cynipoid Phylogenomics
We studied gall-wasps to try to understand the genetic basis of their biology. Gall-wasps
(or Cynipids) are a family of parasitic wasps that attack oaks and other plants. They are
notable for their habit of manipulating their host by forcing it to produce a complex structure
where the larvae are fed and protected. However, some cynipids are unable to initiate the
process and usually live in the gall of other insects (they are hence termed the "inquilines").
In order to identify genes responsible for gall-induction and clarify its exact mechanism, we
compared the genomes of gall-inducers and inquilines. We also worked on improving our
understanding of both the phylogeny of the group and the life-history of the various species,
as this kind of knowledge is essential to the form.
Involved Researchers: Erik Gobbo
Phylogenetic Probabilistic Programming Languages (PhyPPL)
PhyPPL is a Marie-Curie-financed project aimed to achieve the first large-scale application
of probabilistic programming in phylogenetics. It is closely linked to the development of the
TreePPL probabilistic programming language for phylogenetics.
The project's goals are to develop evolutionary models taking advantage of the advanced
inference methods provided by sequential Monte Carlo algorithms and implement them in the new
TreePPL programming language.
The project is supported by a grant from the European Union's Horizon 2020 research and
innovation program under the Marie Skłodowska-Curie grant agreement PhyPPL No 898120 to
Viktor Senderov.
Involved Researchers: Viktor Senderov
 Dipteran taxonomy and morphology
Dipteran taxonomy and morphology
The projects were centered on exploration of the large, "open-ended" genus Megaselia
(Diptera: Phoridae). Large, diverse groups like Megaselia can be excellent tools for
investigating ecosystems but the sheer size and diversity of these groups pose a number of
logistic challenges. The projects focused on the identification of Megaselia in Sweden
(including the description of species new to science), the phylogeny of Megaselia,
streamlining species discovery using massive parallel sequencing (MPS), and recovering life
history data of the clade using MPS.
Involved Researchers: Emily Hartop
 Automatic organism identification using machine learning
Automatic organism identification using machine learning
Rapid and reliable identifications of organisms are important in many contexts, from the
detection of disease vectors and invasive species to the sorting of material from biodiversity
inventories. Because of the shortage of adequate expertise, there has long been an interest in
developing automated systems for such tasks. Today, sophisticated AI models can help us to
automate some of these processes. Although not always at the human expert-level, these models
are doing it much faster and can be applied on scales unmanageable for manual processing.
Our research mainly focused, but was not limited to, the use of state-of-the-art machine
learning in computer vision, convolutional neural networks (CNNs). Our research addressed
questions such as:
- automated taxon identifications using effective feature transfer (paper)
- tackling tasks which are deemed impossible to solve by humans (submitted)
- engaging citizen scientist to effectively collect data (read more about our citizen science
project which engaged 15 000 swedish pupils -
website)
- the use of genetic data for improving image based automated identification tools (in progress)
- the use of CNNs for inferring phylogeny (planned)
- development of ready-to-deploy solutions - for example identification of wildlife from
camera trap images (paper)
Involved Researchers:
Miroslav Valan,
Allison Hsiang
 Metabarcoding of insects for accelerated taxonomic discovery
Metabarcoding of insects for accelerated taxonomic discovery
This project, part of the European project BIG4 consortium
(Horizon 2020 framework - Marie Curie innovative training network), was focused on the
optimization of protocols for metabarcoding of insects from bulk samples or environmental DNA
(eDNA).
Our research stressed the importance of the marker choice as one of the key factors in the
metabarcoding workflow, in terms of maximising taxonomic coverage and resolution, as well as
which marker is more adequate for different types of samples.
We also focused on laboratory methods for non-destructive DNA extraction, trying to find the
best protocol that effectively extracts DNA while minimizing the effect on the structural
integrity of the specimens, so that their morphology can be examined by taxonomists after
obtaining the sequencing results.
This synergy between metabarcoding and taxonomy can speed up the discovery and description of
new species.
Involved Researchers: Daniel Marquina
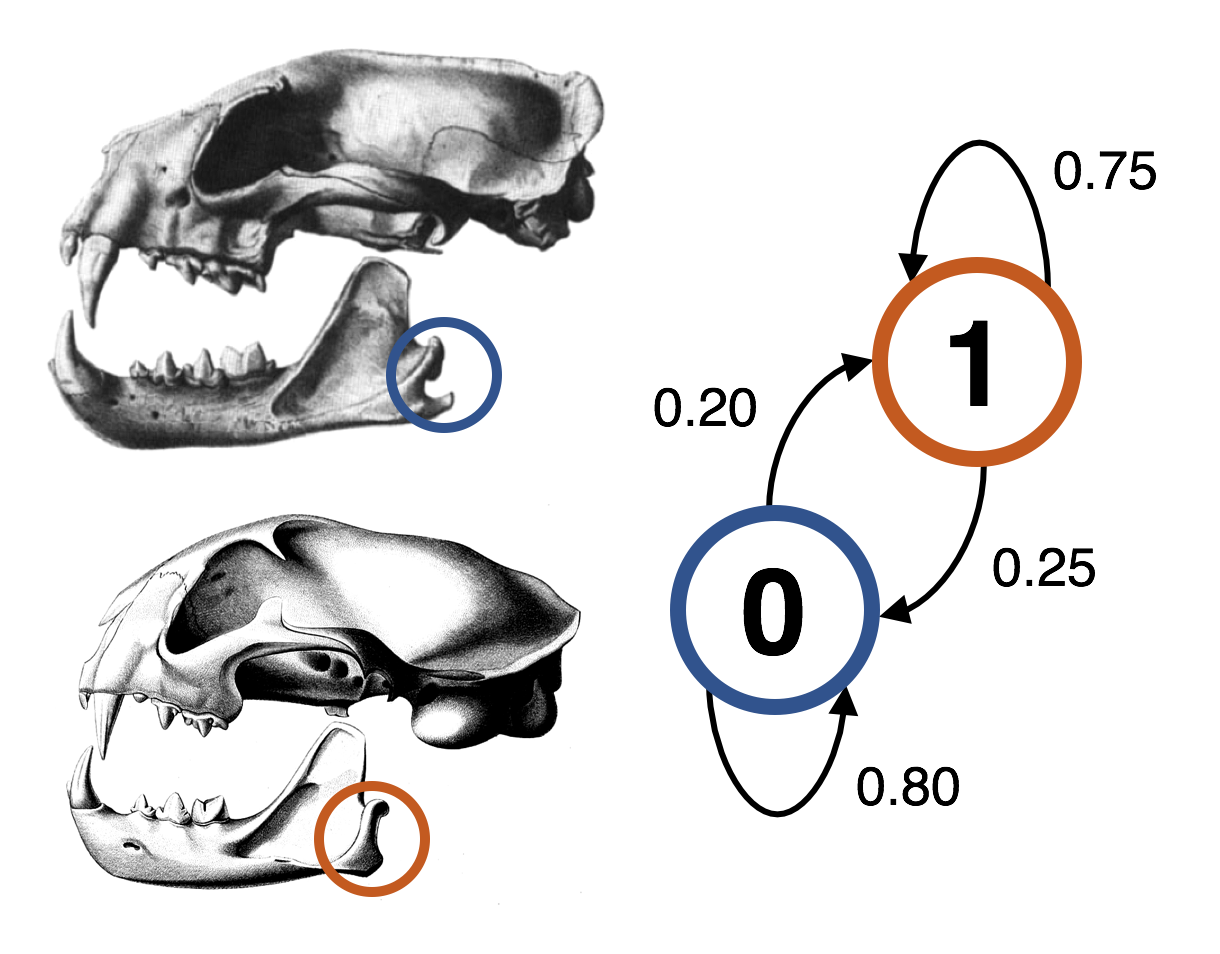 Probabilistic modelling of morphological evolution
Probabilistic modelling of morphological evolution
Morphology represents an important source of data for understanding evolutionary history,
particularly for fossil taxa, which remain the only means by which we can directly observe the
history of life. Modern statistical phylogenetic inference methods using Bayesian and maximum
likelihood frameworks require probabilistic models of evolution, such as the
Generalized Time-Reversible (GTR) model of DNA evolution. However, existing probabilistic
models of morphological evolution are relatively simple, and fail to capture the complexity of
morphological evolution – in the context of phylogenetic inference, their use is analogous to
using only the Jukes-Cantor model for phylogenetic inference using nucleotide data. We are
thus developing new probabilistic models of morphological evolution to address this gap. In
particular, we are modelling correlation between traits, using both discrete-state
continuous-time Markov models and multivariate normal models to detect correlation structure
in phenomic datasets.
Involved Researchers: Allison Hsiang
 Evolution of butterfly-plant associations
Evolution of butterfly-plant associations
In collaboration with Michael Landis (Washington University in St. Louis), we developed a
Bayesian approach for inferring the history of association between butterflies and their host
plants. We describe the evolution of the repertoire of hosts that a butterfly uses as a
continuous-time Markov process of gain and loss of discrete hosts, where gains are influenced
by the phylogenetic distance between hosts. This method will allows us to reconstruct ancestral
networks and investigate more realistic problems that involve ecological interactions among many
taxa. Related papers: Braga et al. 2020 Syst Biol, Braga et al. 2021 Ecol Let.
Involved Researchers: Mariana Braga
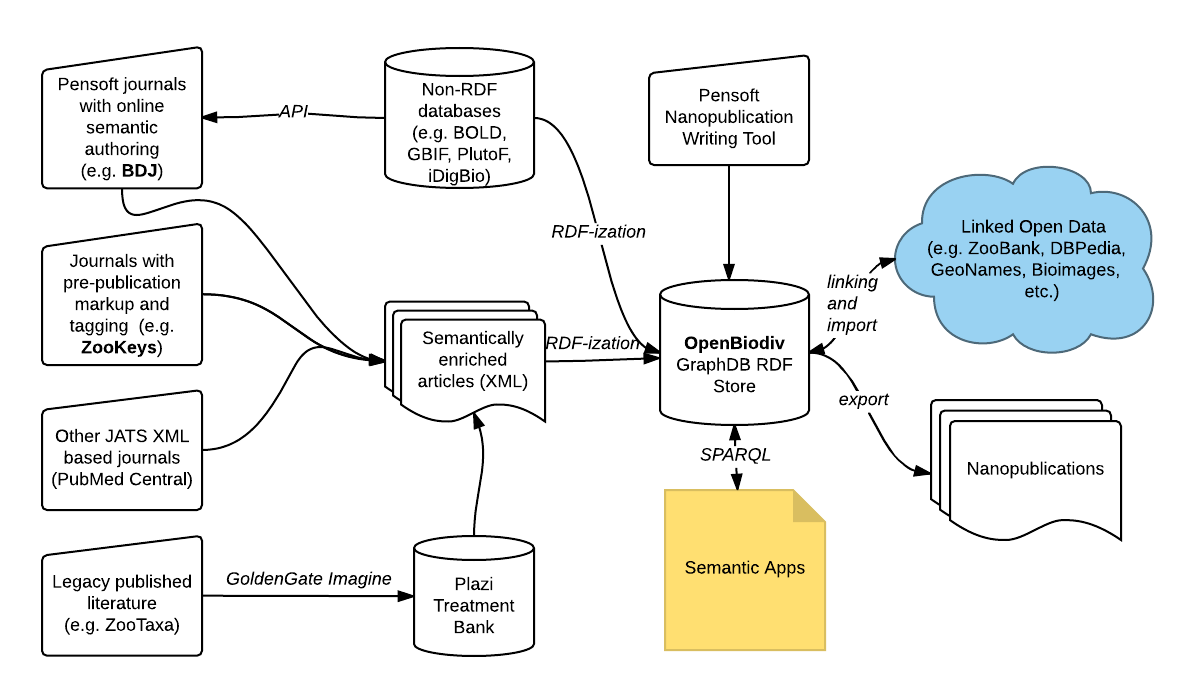 Semantic Knowledge Bases for Biodiversity
Semantic Knowledge Bases for Biodiversity
A knowledge base (KB) about biodiversity called
OpenBiodiv. The KB consists of semantic
apps (expert apps) that answer specific questions that biodiversity scientists have, as well
as general access services running on top of a semantic graph database. The database is
powered by the OpenBiodiv Ontology and populated by information extracted from scholarly
articles.
Involved Researchers: Viktor Senderov