The Ronquist Lab
Computational biology and insect diversity
The Ronquist lab is an interactive and interdisciplinary environment, where we focus on innovative research at the interface between statistics, computer science, evolutionary biology, and insect diversity. We have co-authored some of the leading software packages for Bayesian analysis of phylogenetic problems, and continue to develop new computational approaches, models and inference strategies. The current focus is on universal probabilistic programming. In our empirical research on insect diversity and evolution, we also strive to explore new directions. Recent examples include deep learning for automated image-based identification, improved methods for finding genes linked to life-history changes, more efficient molecular techniques for rapid species discovery, and genetic analyses that scale so that we can analyze the composition and function of entire insect faunas and their associated microbiomes.
News

SIMS volunteer weekend on Öland, March 2026
2026-03-21
In early spring, the volunteers of the Swedish Insect Monitoring Scheme (SIMS) meet the team at Station Linné.
The sampling bottles from last season are brought in, new material is distributed and news from research shared.
Great to see everyone again, and thanks to the volunteers for their amazing contributions of insect samples!

Mistra FinBio annual meeting took place in September 2025
2025-09-25
End of September, the annual Mistra FinBio meeting took place at Näsby Slott in Stockholm for three full days or interdisciplinary science and networking. From the Ronquist lab, Emma, Fredrik and Rob were present to
represent Work Package 1. Rob is otherwise now at the School of Applied Sciences, University of the West of England in Bristol.
Participating institutions: the Swedish Museum of Natural History, Stockholm Resilience Centre, Oxford Sustainable Finance Group, University of Gothenburg, and several from the FinBio Board and Impact Partners.

Lab autumn get-together
2025-09-20
Changseob Lim has returned to the Ronquist lab for a 18 month period as a postdoc! To celebrate and catch-up, the Ronquist lab had a joint lunch and fika at Fredrik's place.

Europe-wide insect monitoring Malaise traps and metabarcoding?
2025-05-06
A group of researchers, specialists and coordinators from 12 EU countries got together in Station Linné on Öland to work on a proposal for Biodiversa+ pilot project. The aim of the project is to harmonize and standardize insect monitoring efforts across Europe. It was great fun to meet in person and work together on the proposal. If successful we will meet again in 2026!

First DarkTree project retreat took place on Öland!
2025-03-19
DarkTree is a KAW-funded initiative to sequence 3,000 insect genomes from poorly known lineages ("dark taxa"). The project integrates DNA metabarcoding, computer vision, and cutting-edge genomic methods to illuminate neglected branches of the insect tree of life.
It is a collaboration between Swedish Museum for Natural History (PIs: Fredrik Ronquist and Tom van der Valk), Linköping University (PI: Michael Felsberg) and KTH Royal Institute of Technology (PI: David Broman).

Mistra FinBio annual meeting took place in September 2024
2024-09-26
End of September, the annual Mistra FinBio meeting took place in Stockholm where we gathered all program members to learn from each other and push forward on the programme's research agenda.
Two full days of workshops, presentations and a bit of field work focusing on environmental DNA sampling and biodiversity monitoring.
Participating: the Swedish Museum of Natural History, Stockholm Resilience Centre, Oxford Sustainable Finance Group, University of Gothenburg, IVL, and several from our Board and Impact Partners.

Gold medal for exceptional scientific work
2023-12-03
Fredrik has received a Memorial Medal in Gold by the Royal Physiographic Society of Lund. The medal is awarded for exceptional scientific work.
The discipline for the award shifts between Biology, Mathematics/Physics and Medicine and is awarded every third year.
The specific motivation was:
"For your research in systematic biology in a broad sense, primarily focusing on the systematics, taxonomy, and biogeography of wasps, as well as the development of the software MrBayes and RevBayes, which are now the standard tools worldwide for phylogenetic reconstruction."
Postdoc in probabilistic machine learning
2023-11-08
We are looking to hire a postdoctoral researcher in probabilistic programming and probabilistic machine learning for problems in evolution and biodiversity.
The lab has a long track record of developing advanced statistical analysis software for phylogenetics, evolution and biodiversity research.
Application deadline: December 3.
Read more about the position and apply here.
Ronquist Lab at Meno Male
2023-11-07
PhD student Alex Reiss from the Stone group in Edinburgh is currently visiting Mariana Braga and the Ronquist Lab.
A good reason to revisit Meno Male for great Italian pizza. We finally got a table, and celebrated recent developments:
papers accepted, papers rejected, funding applications sent in, and medals won for outstanding scientific achievement!
TreePPL preprint available on bioRxiv
2023-10-13
The Ronquist Lab has for several years been involved in developing a universal probabilistic programming language specifically targeting
problems in statistical phylogenetics. Now the preprint describing the TreePPL platform is live on bioRxiv.
This is a highly collaborative effort between computational biologists, computer scientists and evolutionary biologists
at KTH Royal Institute of Technology, BI Norwegian Business School, Université Claude Bernard Lyon 1, the Swedish University of Agricultural Sciences and the Swedish Museum of Natural History.
The general goal is to separate model specification from the implementation of the inference machinery through universal probabilistic programming.
Read more in the preprint and on the TreePPL website.
Optimizing insect metabarcoding using replicated mock communities - paper out now
2023-02-18
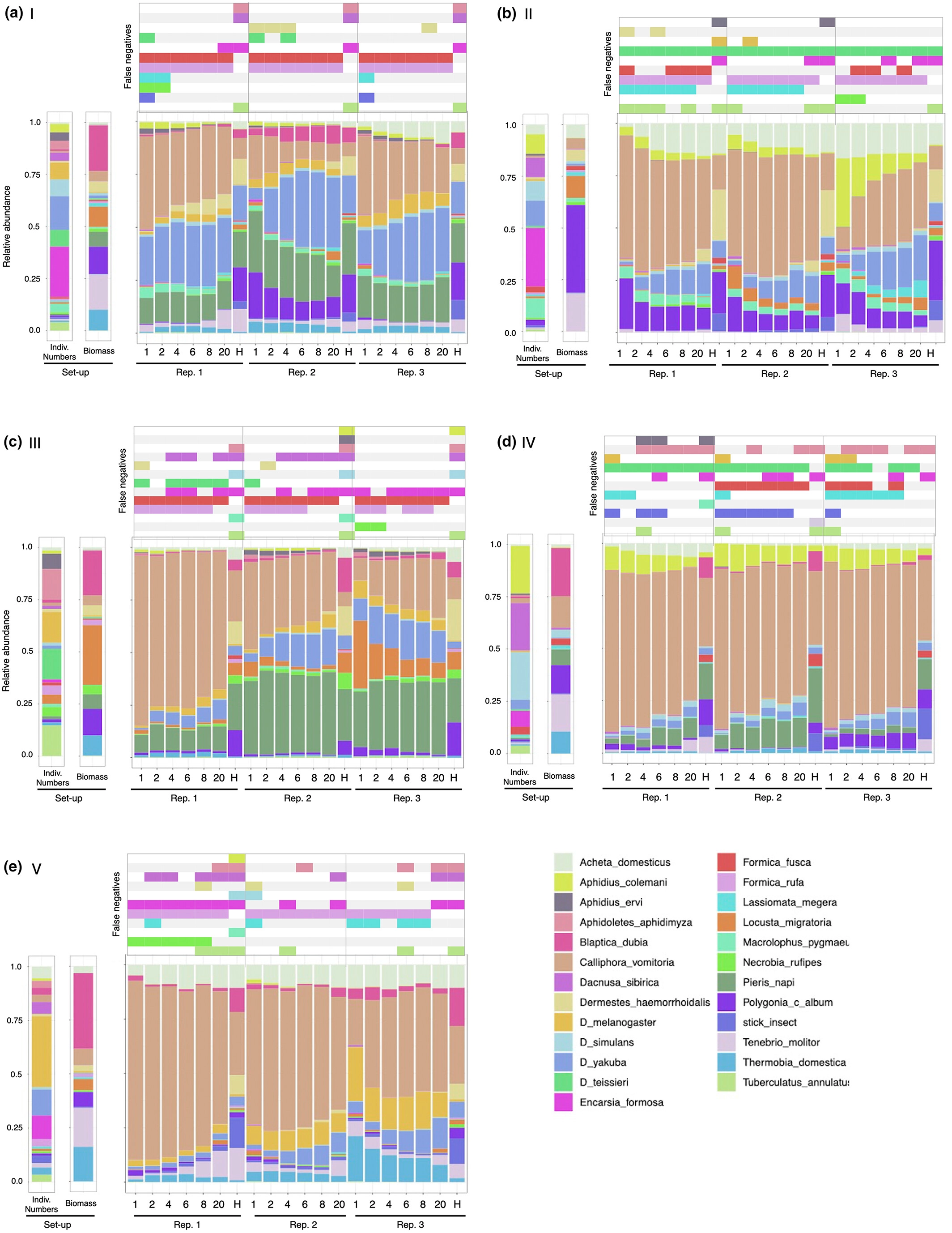
Another milestone on the road to choosing the optimal metabarcoding protocol for bulk insect samples has been reached - paper is out now in Methods in Ecology and Evolution.
We used a series of mock community experiments and a probabilistic model to gain new insights into the methodological choices that determine the reliability of species detection as well as abundance and biomass estimations in the metabarcoding of multi-species community samples. The study is the first to examine variation between biological replicates of multi-species communities and, as such, it provided an unprecedented insight into the extent and significance of variation inherent to metabarcoding. Our results reveal a surprising amount of variation among replicates of identical mock communities. It concerns both the relative read abundance and the presence/absence of species. It was particularly strong after mild lysis treatment but also persisted, to a lesser extent, after homogenization. We demonstrated that a non-destructive, mild lysis approach shows the highest promise for the presence/absence description of the community, while also allowing future morphological or molecular work on the material. However, homogenization protocols perform better at characterizing community composition, in particular in terms of biomass. Small insects are more likely to be detected in lysates, while some tough species require homogenization to be detected.
This work was a close-knit collaboration with Symbio group from Jagiellonian University in Krakow.
Fredrik reaches 100,000 citations
2023-02-08
According to Google Scholar, Fredrik's work has now been cited more than 100,000 times. A lot of the citations are due to MrBayes, which remains widely used for Bayesian phylogenetic analysis. "The popularity of MrBayes is due in part to good timing, it was one of the first tools for Bayesian phylogenetics. But part of the reason, I hope, is that the software has also been fairly easy to use", says Fredrik. "But some of my other contributions have received some attention too", Fredrik is quick to add. Fredrik celebrated by offering the rest of the lab a home-made Swedish "Princess cake", decorated appropriately for the occasion.

IBA team in Madagascar
2023-01-19
The Insect Biome Atlas Stakeholders Briefing meeting "Developing a future monitoring program for Malagasy biodiversity" took place on the 18th of January 2023 at the Madagascar Biodiversity Center. The team travelled to Madagascar to share the first results of the metabarcoding of ca 1700 insect community samples collected by more than 100 local assistants at 50 sites over 12 months, the largest insect material ever sequenced from the tropics. Results show that each National Park has its own unique fauna, with only little overlap with other parks, highlighting that the loss or degradation of even a single park would result in a catastrophic loss of species. We show that by working in close collaboration with local stakeholders it is possible to bring into the realm of biodiversity monitoring such highly diverse and previously-unknown parts of Malagasy fauna. There is still much work ahead for the IBA team, but we came back from our trip to Madagascar full of energy and inspiration to get the data out and share these exciting results (and many others) with the wider scientific community.
Automatic alignment at ESOP 2023
2023-01-08
When working on probabilistic programming for diversification models, we discovered the importance of "alignment". In short, when you apply inference strategies to probabilistic programs, you need to pay attention to how different simulations from the model match each other at intermediate points, or else the inference can become very inefficient. Previously, we used manual alignment of probabilistic programs to address this but, under the lead of David Broman's group at KTH and his student Daniel Lundén, we have now developed automatic alignment. This work will appear at ESOP 2023 in Paris, April 22-27.

Erik Gobbo nails his thesis at the Stockholm University
2022-10-05
As required by the Stockholm University's tradition, all PhD theses need to be nailed in a public place before the defence so that the wide audience can read it and comment. Today Erik Gobbo nailed his thesis titled "Gall induction n gall wasps (Cynipidae s. lat.) - Insights from comparative genomics" at the Department of Zoology.
The defence will happen on the 26th of October 2022 at 9h in Vivi Täacholm room.

Optimizing protocols for the non-destructive extraction of DNA from insect community samples
2022-09-01
One of the group's goal, to make use of massive molecular identification methods (DNA metabarcoding) to facilitate and accelerate taxonomic work in the most optimal and efficient way, usually conflicts with the taxonomic work itself. This is because most protocols for metabarcoding require the destruction of the insects to better extract the DNA from them, making it impossible to conduct any morphological examination on the individuals. In a paper recently published in Metabarcoding & Metagenomics, we continued exploring and optimizing protocols for the non-destructive extraction of DNA from samples with a mixed community of insects. In this study we tested different lysis buffers, incubation times, and post-lysis purification of the DNA, and how these factors affected not only the metabarcoding output, but also the preservation of the morphological features of the insects.
We found that the most optimal protocol consists of a mildly aggresive lysis buffer (low concentration of proteinase K, without other proteolytic compounds, lower concentration of membrane-breaking surfactants) and a short incubation time, regardless of whether a manual and labour-intensive purification or an robot-automatic one is used. This combination not only maintained better the morphological characteristics of the insects, but also produced the most accurate estimations of community composition, compared to the other protocol combinations. Fortunatley, this is also the cheapest (lower amount of reactives needed) and fastest of the tested options!
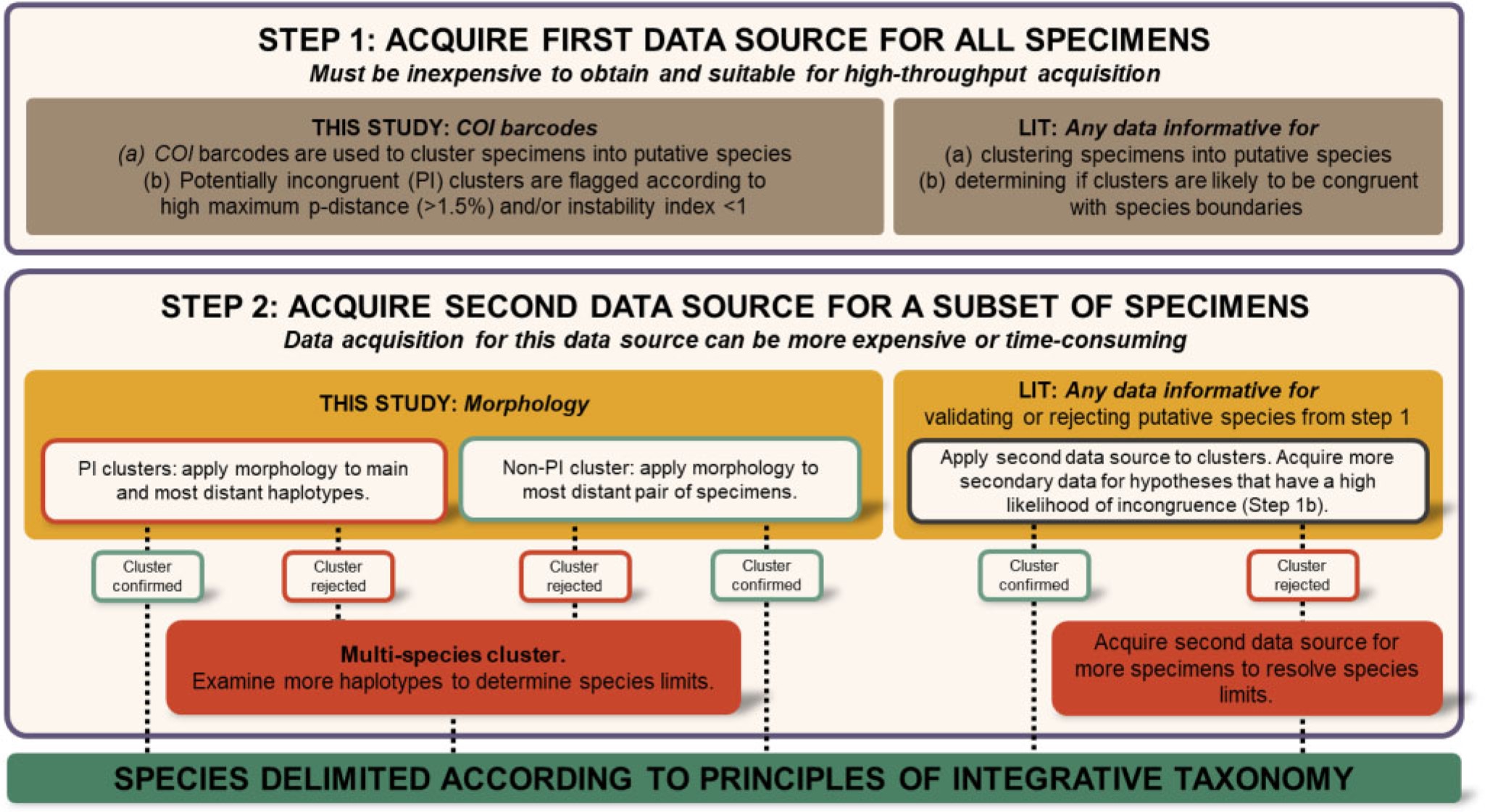
Large-scale integrative taxonomy (LIT) paper is out
2022-07-21
Large-scale integrative taxonomy (LIT) is a new, rapid, systematic and objective species delimitation method specifically designed for tackling "dark taxa". Dark taxa are defined in the paper as groups for which <10% of all species are described and the estimated diversity exceeds 1,000 species. Due to the sheer numbers of species and specimens in these groups, coupled with the complexity of both molecular and morphological species determination, multiple data sources must be used to delimit species ("integrative taxonomy"). To make collecting this much data an efficient and scalable practice, LIT relies on preliminary species hypotheses that are generated based on inexpensive data (in the paper, COI barcodes) that can be obtained quickly and cost-effectively. These hypotheses are then validated using a more expensive type of data that is only obtained for specimens selected based on objective criteria applied to the preliminary species hypotheses (in the paper, morphology). The paper developed and tested LIT using a dataset of 18,000 scuttle flies (Diptera: Phoridae) that were first grouped into 315 putative species based on barcode data. Using LIT, approximately 1000 specimens (6% of total) were selected for morphological examination. LIT reveals that it is possible to predict the molecular clusters likely to be incongruent with species boundaries, and to objectively pick a small subset of specimens to use for validation.

Emily Hartop's thesis defence: A multifaceted approach to a "dark taxon"
2021-06-08
On the 8th of June Emily Hartop successfully defended her PhD thesis: A multifaceted approach to a "dark taxon". A dark taxon is a hyperdiverse yet understudied taxonomic group.
Her work focused on the scuttle flies (Diptera: Phoridae), including for example a comprehensive molecular phylogenetic analysis on the scuttle fly genus Megaselia, as well as method developments for large-scale species discovery and description.
Emily is now a researcher on Hyperdiverse Diptera at the Museum für Naturkunde in Berlin.
Universal probabilistic programming offers a powerful approach to statistical phylogenetics
2021-02-24
In a paper appearing online today in Communications Biology, we outline our recent work in probabilistic programming,
and demonstrate that universal probabilistic programming languages (PPLs) solve the expressivity problem that exists in phylogenetic analysis, while still supporting automated generation of efficient inference algorithms.
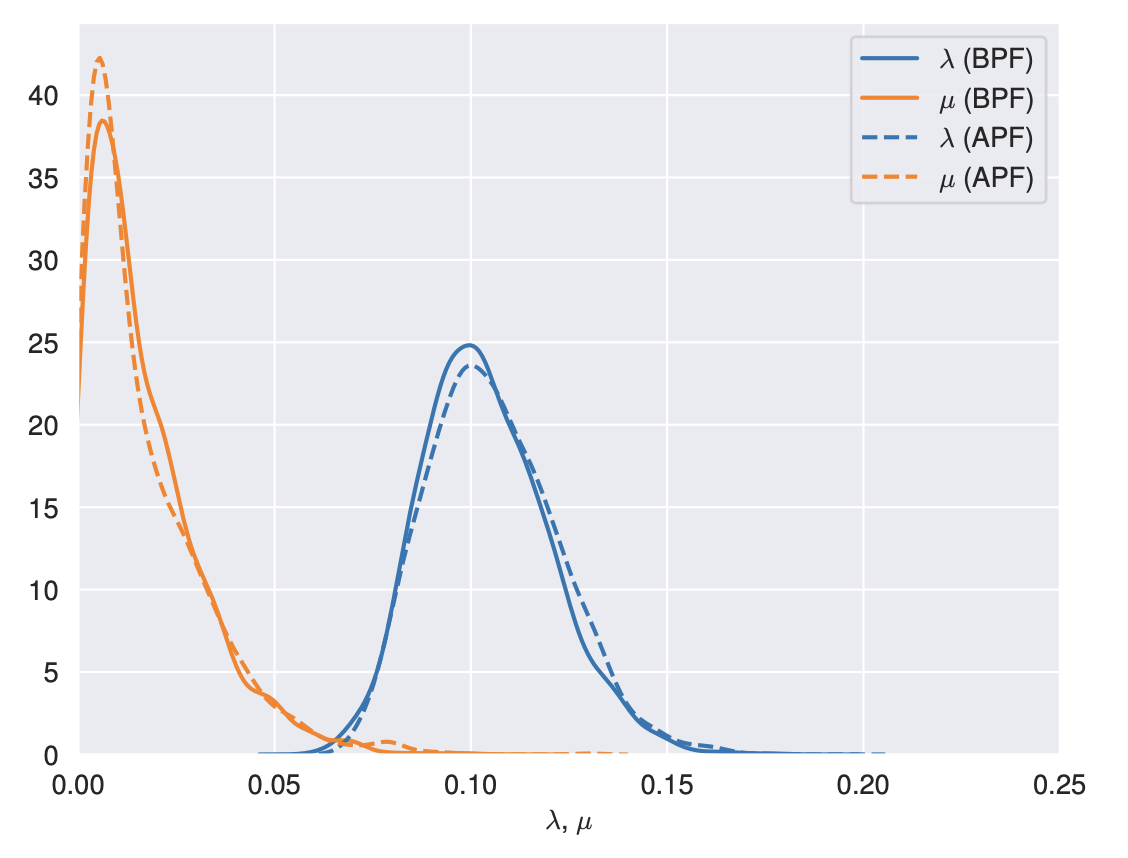
The paper presents automated generation of sequential Monte Carlo (SMC) algorithms for PPL descriptions of arbitrary biological diversification (birth-death) models. Applying these methods to 40 bird phylogenies we show that models with slowing diversification, constant turnover and many small shifts generally explain the data best.
The field of probabilistic programming is currently in a phase of experimentation, and few hurdles remain before these techniques can be effectively applied to the full range of phylogenetic models.

Effects of ethanol concentration on insect preservation
2021-02-12
In a paper published today in PeerJ, we study the effects of ethanol concentration on the preservation of insects. High concentrations of ethanol are widely believed to make insects brittle, justifying the preservation of insect specimens for morphological study in 70-80% ethanol. However, recent taxonomic and phylogenetic research is usually based on DNA sequencing, and DNA is better preserved at much higher concentrations. This causes a trade-off between morphological and molecular preservation that has rarely been studied to date.
We analyzed the effects of different ethanol concentrations on morphological and molecular preservation in seven insect species. We found that robust species suffer little morphological damage at high ethanol concentrations, and there is noticeable degradation of their DNA at intermediate to low concentrations. In contrast, weakly sclerotized species were susceptible to damage in high concentrations of ethanol, but their DNA was preserved well also in intermediate concentrations of ethanol. Interestingly, at concentrations of around 90%, the preservation of both morphology and DNA was acceptable for most species.
The paper is the result of a collaboration between our lab and the Symbiosis Evolution group at the Jagiellonian University in Krakow.

Royal Swedish Academy of Sciences Awards Prize to Dave Karlsson
2021-02-11
On February 11, the Royal Swedish Academy of Sciences announced that they were awarding the Sture Centerwall Prize to Dave Karlsson, PhD student in the Ronquist lab and Manager of Station Linné, for his dedicated and tireless work in documenting the Swedish insect fauna, and in reaching out to the general public and raising their awareness of insect diversity and the importance of insect conservation.
Read more about the award on the Academy web site here.
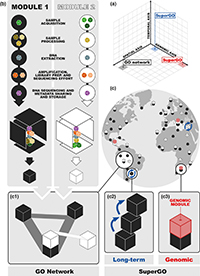
Genomic Biodiversity Inventories
2021-01-12
High-throughput sequencing methods are revolutionizing biodiversity monitoring. In a perspective piece published today in Molecular Ecology, resulting from a November 2019 workshop in Cyprus, we contribute to recommendations for how current and future projects using these new approaches to study biodiversity can be more effectively connected to provide a site-based genomic framework for global integration and synthesis.
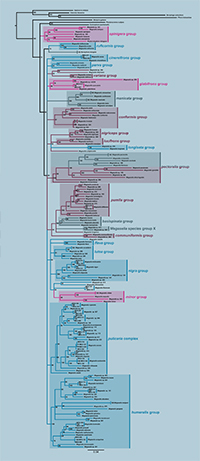
Phylogeny of scuttle flies
2021-01-09
The scuttle fly genus Megaselia is one of the most species-rich in the animal kingdom, with roughly 1,700 described species and probably at least an order of magnitude more species remaining to be discovered. In large part because of the enormous diversity, phylogenetic relationships within the genus are still very poorly known, complicating further taxonomic work on the radiation. In a paper appearing online today in Systematic Entomology, we present the first comprehensive molecular phylogenetic analysis of relationships within the genus.
We find that most of the species diversity of Megaselia is found within a "core" clade. Future restriction of the genus to this lineage would render it monophyletic but it would necessitate the recognition of a few lineages outside of the core lineage as separate genera.
Within the core Megaselia, we identify twenty-two well-supported species groups. This establishes an important phylogenetic scaffold based on DNA sequence data that will facilitate future exploration of the radiation. Nevertheless, many relationships within the genus will continue to be poorly resolved until it becomes possible to significantly expand both the species representation and the amount of sequence data.
Postdoc in probabilistic programming
2020-11-24
We are looking for a postdoc interested in developing new modeling and inference tools based on universal probabilistic programming, an approach that has attracted considerable attention across scientific disciplines in recent years. Specifically, we will be developing a domain-specific language to describe phylogenetics problems, and design new inference strategies for such model descriptions. The goal is to successfully tackle some of the most challenging research problems in statistical phylogenetics and phylogenomics. For some early success stories, see our recent paper on bioRxiv. The postdoc will be encouraged to develop new projects, apply for third-party funding, supervise students and lecture at courses. Read more and apply here. Application deadline December 18.

Unique insect data published
2020-11-24
The Swedish Malaise Trap Project (SMTP) is q unique contrywide inventory of a national insect fauna. The field campaign was completed in 2003-2006, and sampled the insect fauna at 73 different selected sites across the ccountry using Malaise traps. The entire catch is estimated to contain some 20 million insect specimens. This unique project is now described in a recent paper of ours in Biodiversity Data Journal. More than 100 taxonomic experts have participated in the identification of the material so far. Around 170,000 specimens, about 1 % of the total material, have been determined to species. What is unique about these data is that they are focused to a large extent on poorly known groups in the insect orders Hymenoptera and Diptera, groups that are rarely processed in similar inventories for lack of taxonomic expertise. The available SMTP data have recently been published to GBIF as 79 sample-based datasets, as described in another recent paper of ours, also in Biodiversity Data Journal.
Thanks in large part to the SMTP inventory, the known Swedish insect fauna has increased with 2,000 species in the last decade, to 28,000 species. Our recent analysis based on the inventory data suggests that as many as 5,000 additional insect species may still remain to be discovered in Sweden, many of them likely new to science. Most of the remaining species belong to Hymenoptera and Diptera, and many of them are decomposers or parasitoids.
2019 Publisher's Award for Excellence in Systematic Research Awarded to Miroslav
2020-11-16
We are proud to announce that Miroslav Valan, one of the PhD students in the lab, was one of two winners of the 2019 Publisher's Award for Excellence in Systematic Research. The award is presented to the two best papers based on student research published in Systematic Biology during the previous year. The lead author must have been a student at the time the research was conducted. Miroslav won the prize for his paper developing methods for automated image-based identification of insects based on convolutional neural networks and feature transfer.

Hakuna Ma-Data Microsoft Sponsored Competion Won by Miroslav
2020-11-16
Miroslav Valan, one of the PhD students in the Ronquist lab, recently won the Hakuna Ma-Data competition sponsored by Microsoft. The task was to automatically identify species of wildlife in camera trap images from the Serengeti National Park. His contribution placed first among contributions from over 800 teams, earning him a $20,000 contribution to his research. Miroslav's solution was presented at the prestigious computer-vision conference CVPR 2020.